Computational Drug Discovery Part 2: Molecular Representations for Machine Learning
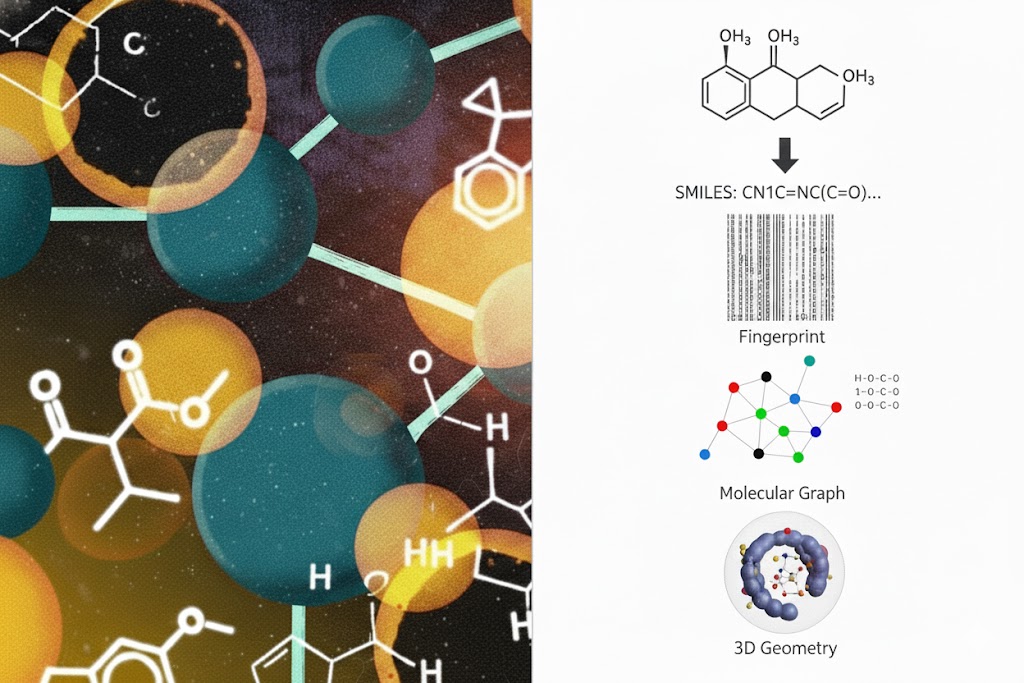
Molecules are converted into data for ML using SMILES, fingerprints, molecular graphs, and 3D geometry. The trade-off is between compactness (SMILES/fingerprints) and richness (graphs/3D geometry). The representation choice fundamentally determines what models can learn about binding and performance in drug discovery.